Cell-free or Genomic DNA-Based mNGS Assays: What’s Best for Infectious Disease Diagnosis?
Study Shows gDNA Better Suited for Infectious Disease
Study Shows gDNA Better Suited for Infectious Disease
Micronbrane Medical sets a new standard for mNGS-grade reagents to enhance the accuracy of pathogen identification.
Infectious diseases claim over 18 million lives each year.¹
Finding the root cause of infections is urgent as current testing methods fail to identify the causal agent up to 60% of the time.² This crisis leaves a massive gap in effective treatment with negative downstream effects such as the emergence of antimicrobial resistant and multidrug resistant organisms plus complications that extend hospital stays and drive up healthcare costs. Microbiologists are seeking a rapid, accurate, and affordable way to identify pathogens and metagenomic Next-Generation Sequencing (mNGS) could be a game-changer.
In many cases, mNGS is not just an option but the only hope to identify complex microbial infections. mNGS offers an unbiased and hypothesis-free approach, capable of identifying known, novel, and emerging microbes. Its ability to detect low-abundance microorganisms and antimicrobial resistance genes, while offering comprehensive insights into microbial functions and host response, has transformed our understanding of the vast microbiome.
However, amidst the promises of mNGS lies a formidable challenge – the contaminome. As microbes constitute only a tiny fraction of genomic material, host DNA and nucleic acid contamination often obscure microbial signals, leading to decreased detection sensitivity, increased workflow complexity, and slower clinical adoption. The contaminome casts a shadow over the potential of mNGS, hindering its widespread implementation, especially in clinical settings.
To shed light on the impact of the contaminome in mNGS, we’ve crafted a fictional narrative titled “Conquering the Contaminome.” In this tale, the contaminome emerges as the villain, lurking everywhere in the mNGS workflow, shedding nucleic acids, and confounding results.
At Micronbrane Medical, our strategic focus is on “conquering the contaminome” with an all-in-one, accelerated, affordable, and automatable assay PaRTI-Seq™ and bioinformatic pipeline PaRTI-Cular™, empowering researchers and clinicians with a ‘superpower’ to transform the study, diagnosis, and treatment of infectious diseases. This journey begins with the first of many episodes available at micronbrane.com/contaminome.
Join us in our quest to conquer the contaminome and unlock the full potential of mNGS. Together, we can pave the way for a healthier, more resilient world.
1-2. World Health Organization

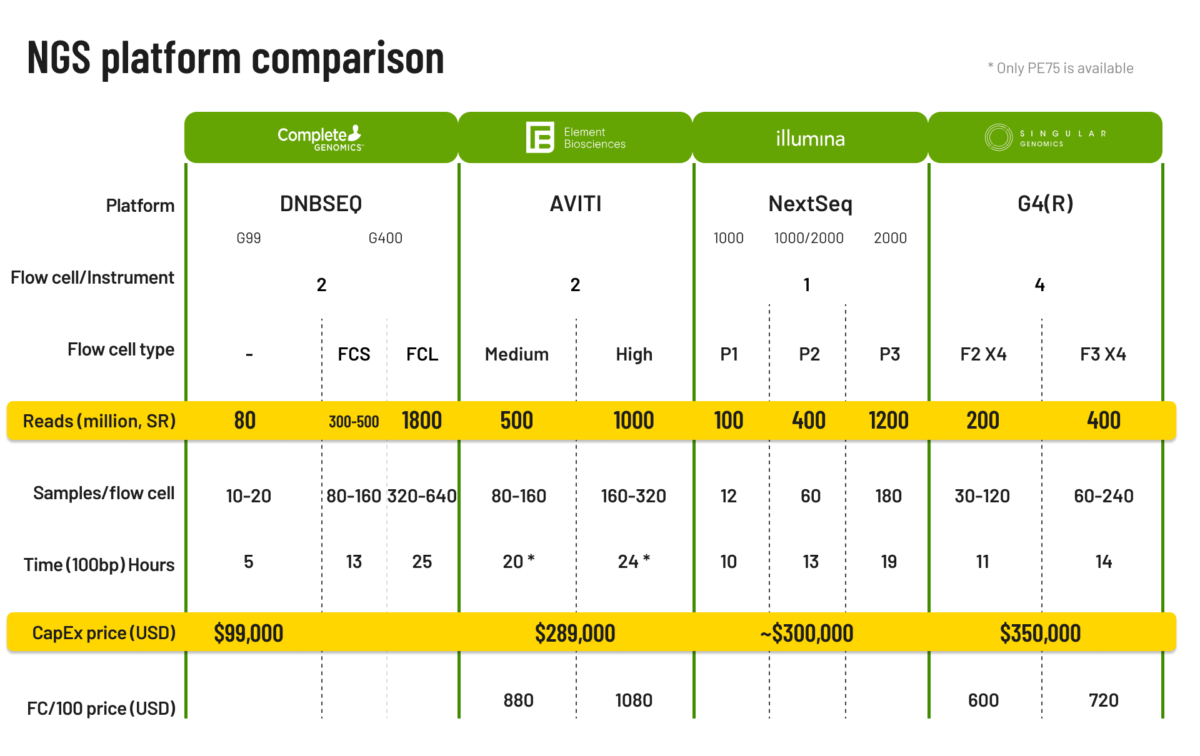
Inquiries about short read sequencing platforms for mNGS applications are common among our customers. While the answer depends on specific requirements, we’ve conducted an in-depth analysis to aid in your decision-making process.
Sanger sequencing, known as the first-generation method, sequences DNA post-synthesis.
Despite the label “Next-generation Sequencing (NGS),” the advent of sequencing by synthesis technology for commercial use emerged around 2005. For two decades, the market was dominated by Thermo Fisher with Ion Torrent and Illumina, offering a variety of machines. Illumina utilizes a fluorescent tag to identify nucleotide bases added to the DNA strand, while Ion Torrent detects hydrogen release during nucleotide base addition, conducting both sequencing and synthesis concurrently.
Today, there are more short read sequencing manufacturers on the market, from Element Biosciences, Complete Genomics/MGI, to Singular Genomics and Ultima Genomics.
First, we provide a very brief overview of the new entrants then we compare the options for a variety of mNGS applications.
With over a decade of global market presence, this sequencer employs rolling circle replication (RCR) to mitigate clonal errors and index hopping, thereby enhancing accuracy. Conceptually, their rolling nanoball technology enables dual sequencing by synthesis. Their product range and instruments offer diverse outputs suitable for various applications, with enhanced speed due to DNBSeq technology.
This benchtop sequencer tackles pre-facing or facing errors through specialized chemistry. The platform’s patented technology and unique chemistry bolster sequencing read accuracy and quality.
The G4 from Singular Genomics addresses flexibility and scalability needs with the capability to accommodate up to four flow cells simultaneously. This versatility enables users to tailor sequencing setups for different projects or output requirements, optimizing resource allocation.
Ultima Genomics‘ UG 100, a high-output, large-scale instrument, boasts a distinctive feature—no flow cells. Sequencing reactions, synthesis, and signal detection occur on a wafer, accompanied by automation to minimize manual intervention. Their PDM sequencing chemistry enhances accuracy, particularly in detecting single nucleotide variants at low frequencies, beneficial for oncological applications, especially utilizing cell-free DNA with minimal input and mutation frequencies.
For research and for clinical applications the priority of the features are slightly different. For clinical use accuracy, flexibility and turnaround times may be more important than in research settings. We evaluated the benchtop sequencers with medium output that are most appropriate for metagenomic analysis along several dimensions:


Micronbrane Medical’s Pathogen Real-Time Identification (PaRTI-Seq) assay is a cost-effective solution to from collection to action within 24 hours.
However, it’s essential to note that while selecting the sequencer is pivotal, achieving metagenomic goals also hinges on assay selection and the utilization of automation instrumentation. At Micronbrane Medical, we focused on developing technologies that overcome the barriers to ubiquitous adoption of mNGS in all settings. Our Pathogen Real-Time Identification by Sequencing (PaRTI-Seq) assay and our PaRTI-Seq Analysis bioinformatic pipeline are revolutionizing infectious disease study, diagnosis, and tracking from sample collection to definitive action.
AMR occurs when bacteria, viruses, fungi, and parasites change over time and no longer respond to existing treatments. But how does antimicrobial resistance occur and what role metagenomic next-generation sequencing can play in combating it?
Infectious diseases have posed significant health challenges throughout human history. While the advent of antibiotics and other antimicrobials was aimed at addressing this concern, the persistent and widespread use of these agents has led to the emergence of antimicrobial resistance (AMR) and numerous multiple drug-resistant organisms (MDROs).
Let’s get into the nitty-gritty!
In 2019 the World Health Organization (WHO) described AMR as one of the top ten global threats to public health – a threat to which science is playing catch-up in its efforts to mitigate.
Close to 5 million lives are lost annually due to drug-resistant infections.2 Projections suggest that without intervention, global deaths attributable to AMR could reach 10 million by 2050 (the same as cancer deaths).3 In addition to high mortality and morbidity, the economic toll of AMR is substantial. In fact, the World Bank estimates that AMR could result in US$ 1 trillion additional healthcare costs by 2050, and US$ 1 – $3.4 trillion gross domestic product (GDP) losses per year by 2030.4
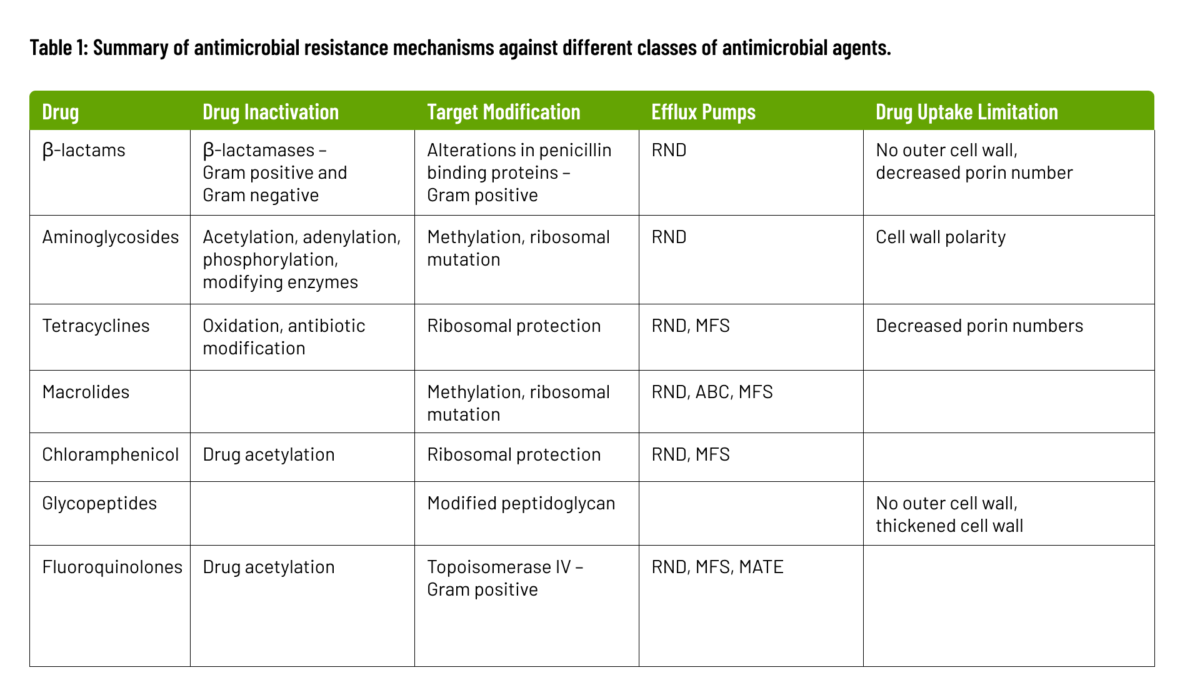
The biochemical and genetic mechanisms that cause AMR, fall into four categories: inactivation of the antimicrobial molecule; target modification; active drug efflux; and drug uptake limitation. These mechanisms are summarized below and in Table 1.
Antimicrobial resistance occurs when microorganisms produce enzymes that deactivate drugs by either destroying them or adding specific chemical components, rendering the antimicrobial ineffective at its target site. These modifying enzymes, catalyzing reactions like acetylation, phosphorylation, and adenylation, induce steric hindrance, diminishing the drug’s affinity for its target and raising the bacterial Minimum Inhibitory Concentration (MIC). β-lactam resistance exemplifies this, employing β-lactamases to break amide bonds in the β-lactam ring, rendering the antimicrobial ineffective. Over 1000 β-lactamases have been identified, with more expected as bacterial evolution continues.
Target modification is a key resistance mechanism involving alterations to the antimicrobial target site, impeding proper binding of the antimicrobial molecule. These sites are crucial for cellular functions during antimicrobial action. Mutational changes on the target site can reduce inhibition susceptibility while preserving essential cellular functions. In some cases, inducing resistance through modifying target structures may require additional cellular changes. An example is the acquisition of penicillin-binding transpeptidase (PBP2a) in methicillin-resistant Staphylococcus aureus.
Efflux pumps, found in families like the major facilitator superfamily (MFS), small multidrug resistance family (SMR), resistance-nodulation cell division family (RND), ATP-binding cassette family (ABC), and multidrug and toxic compound extrusion family (MATE), have the capacity to expel antimicrobial agents rapidly from the bacterial cell. This expulsion mechanism significantly contributes to multidrug resistance.
Bacteria vary in their ability to limit drug uptake. The outer membrane composition in organisms like gram-negative bacteria slows antimicrobial penetration. Mycobacteria’s lipid-rich outer membrane hinders hydrophilic drug entry. Organisms lacking a cell wall, such as Mycoplasma, are inherently resistant to cell wall-targeting agents. Biofilm formation protects against immune system attacks and provides defense against antimicrobial agents.

Summary of Antimicrobial Resistance Mechanisms Against Different Classes of Antimicrobial Agents
Origins of Antimicrobial Resistance
Microorganisms demonstrate genetic plasticity, enabling them to evolve resistance mechanisms against environmental threats, including antimicrobial agents. The development of antimicrobial resistance involves various processes:
Susceptible microbial populations undergo mutations in genes affecting drug activity, facilitating cell survival in the presence of antimicrobial agents.
Random mutation events, arising from replication errors or incorrect DNA strand repairs, contribute to antimicrobial resistance.
Certain bacterial populations enter transient states of elevated mutation rates under prolonged non-lethal antibiotic pressure. Mutators in this context confer selective advantages.
Mutations occur in slowly dividing or non-dividing cells under non-lethal selective pressure, leading to the development of resistant mutants.
Bacterial evolution is influenced by the acquisition of foreign materials through HGT mechanisms, such as transformation, conjugation, and integrons. These processes contribute to the development of antimicrobial resistance.
Metagenomic Next-Generation Sequencing (mNGS) provides unbiased, culture-independent diagnosis and surveillance of resistance mechanisms. Recognized as an indispensable tool, mNGS provides a complete genomic sequence and unparalleled structural detail on individual traits within a population, which contributes to more reliable microbial identification, definitive phylogenetic relationships, and a comprehensive catalog of traits. Additionally, mNGS can also be used for outbreak investigations, microorganism-agnostic infectious disease diagnosis, especially for novel pathogens and appropriate treatment selection.
Understanding the mechanisms and origins of antimicrobial resistance is crucial in developing effective strategies to mitigate its impact. Learn how our Pathogen Real-Time Identification by Sequencing (PaRTI-Seq™) complete mNGS assay provides rapid and accurate identification of resistant pathogens and invaluable insights for the management of AMR.
Sources