Nucleic acid contamination compromises mNGS data analysis which can have alarming repercussions. In fact, a study published in the ASM Journal, “Major data analysis errors invalidate cancer microbiome findings” joins a growing body of evidence illuminating another troubling phenomenon in metagenomic Next-Generation Sequencing (mNGS) — database contamination.
Types of database contamination
More must be done to understand and mitigate the issues with metagenomic data analysis such as:
- Mis-labeling human or host DNA as microbial genomes1
- Sequencing reads mis-aligned to other microbial species2
- Incomplete reference genomes, especially for emerging species
- Errors in MAG construction (QC, assembly, binning or annotations)
- Other computational errors
In metagenomic research, database contamination can ruin study outcomes as in the above paper and many more, it also slows the adoption of mNGS use in clinical settings. What characteristics would high-quality databases have?
High-Fidelity Metagenomic Data Analysis
To avoid the errors that may invalidate findings and allow mNGS to be used in the first line in clinical microbiology, analysis programs, need the following:
- State-of-the-art computational and QC algorithms
- Extensive and ongoing curation of the taxonomic assignments of individual pathogens
- Proper benchmarking of microbes with unusual sequence homologies and/or close taxonomic relationships
- Quick and effective tracking of potentially mis-annotated and/or misrepresented pathogenic species
PaRTI-Cular™ is a bioinformatics web app four years in the making from Micronbrane Medical. The company’s strategic emphasis is on removing technical and cost barriers for the widespread use of mNGS in microbiology. The web application was specifically developed to avoid genome mis-classification, taxonomic irregularities, erroneous variant calls and other fatal errors.
PaRTI-Cular streamlines the analysis of metagenomic Next-Generation Sequencing (mNGS) data using our Pathogen Real-Time Identification by Sequencing (PaRTI-Seq) assay. The web app is a deeply curated, up-to-date genome database for over 1400 microorganisms. The software also automatically conducts data quality checks, removal of host genome reads, background noise cut-offs, pathogen identification. We also developed a proprietary reference database so the platform can deliver abundance distributions and other types of statistical analyses.
With PaRTI-Cular you do not need any programming or special computers to run analyses. In fact, you can upload sample data and get results, from any computer, anywhere in the world. With the RUO version the following functionality is available:
- PaRTI-Cular analysis applies preset parameters developed by Micronbrane Medical using Burrow-Wheeler Aligner (BWA) mapping tools. Mapping parameters such as read length, identity percentages, etc., were tested in various low biomass human clinical samples.
- Data is only mapped to the curated database of 1400 pathogens (no other species are supported at this point.)
- Sequencing output is transfer and PaRTI-Cular analysis uses Micronbrane Medical’s web services hosted by AWS in Singapore
The process is simple: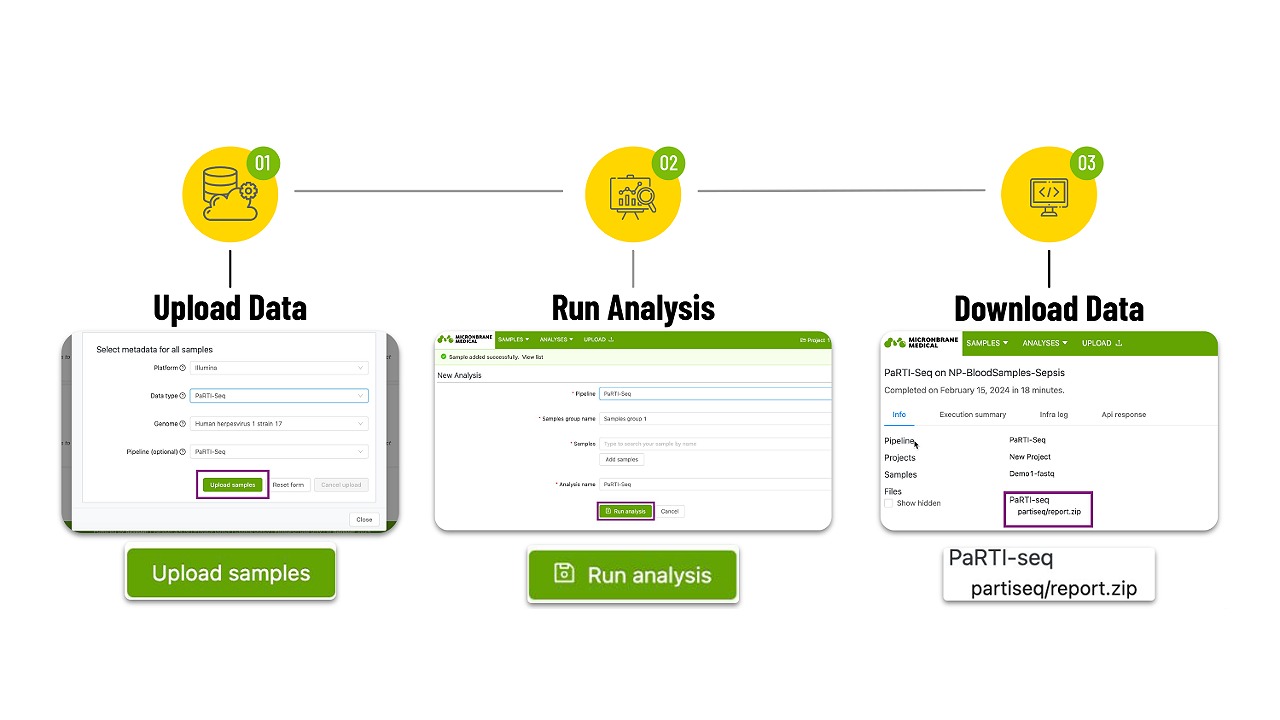
The next PaRTI-Cular launch will include expanded functionality for clinical settings, including:
- Various QC check for run validity and data quality
- Validated cutoff value will be implemented in the pipeline to report potential positives identified from the samples
- Approval process for the final report
- Data and analysis can be housed in user’s own AWS account and data center of selection
Please let us know if you would like to try PaRTI-Cular and be part of its development for both research and clinical use.
Sources
- Breitwieser FP, Pertea M, Zimin AV, Salzberg SL. 2019. Human contamination in bacterial genomes has created thousands of spurious proteins. Genome Res 29:954–960
- Steinegger M, Salzberg SL. 2020. Terminating contamination: large-scale search identifies more than 2,000,000 contaminated entries in genbank. Genome Biol 21:115.